
J-OCTA
J-OCTA
J-OCTA 機能・サービス
J-OCTA(ジェイ・オクタ)は、高分子材料、複合材料、エネルギー材料、エレクトロニクス材料、機能性薄膜・コーティングなどのマテリアルサイエンス分野から、創薬・製剤などのライフサイエンス分野、そして両分野にまたがるバイオマテリアルにも幅広く対応する、マルチスケールシミュレーション・ソフトウェアです。原子・分子からマイクロメートルスケールまでの構造や物性を予測し、複雑な現象の理解や新たな設計指針の創出に役立ちます。シミュレータ同士は共通プラットフォーム上で連携でき、マテリアルズ・インフォマティクスのための機械学習やデータサイエンスとの統合も可能です。研究開発の精度向上や効率化を実現し、マテリアルサイエンスとライフサイエンスの双方でイノベーションを加速させます。豊富な導入実績と手厚いサポートにより、初めての方でも安心してご活用いただけます。
- マテリアルサイエンスのためのシミュレーション
- ライフサイエンスのためのシミュレーション
- マテリアルズ・インフォマティクス
- サポートとコンサルティングサービス
マテリアルサイエンスのためのシミュレーション
材料設計では、ナノからマクロに至る多階層(マルチスケール)構造の理解と制御が不可欠です。J-OCTAは、スケールに対応して量子論から連続体理論までをカバーする統合シミュレーションプラットフォームであり、全原子モデル、粗視化モデル、連続体モデルを駆使し、構造・物性の予測からプロセス設計まで対応しています。力学・熱・電気・光学など多様な物性評価に対応し、スケール間連携やデータサイエンスとの統合も可能です。豊富な事例やチュートリアルにより、初めての方でも安心して利用でき、研究者や技術者が構造や挙動の理解を深め、設計・開発へ応用することを支援します。
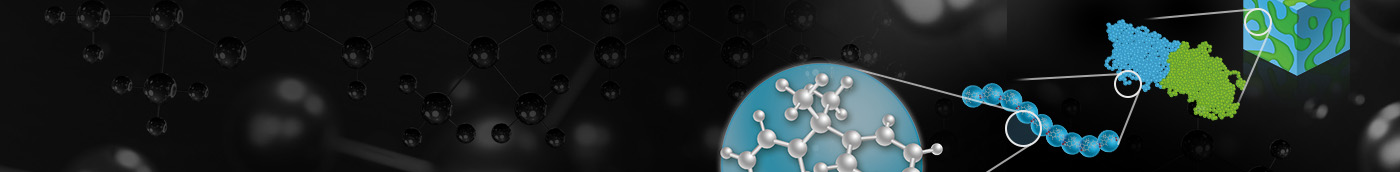
全原子モデリング
全原子モデリング

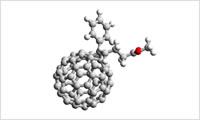
粗視化モデリング

粗視化モデリング

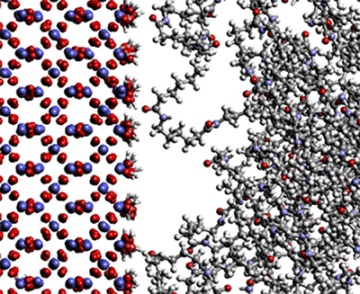
マイクロメカニクス・マイクロ流体・プロセスシミュレーション

マイクロメカニクス・マイクロ流体・プロセスシミュレーション
豊富なシミュレーションエンジン群
豊富なシミュレーションエンジン群
シミュレーション結果の解析と物性推算

シミュレーション結果の解析と物性推算
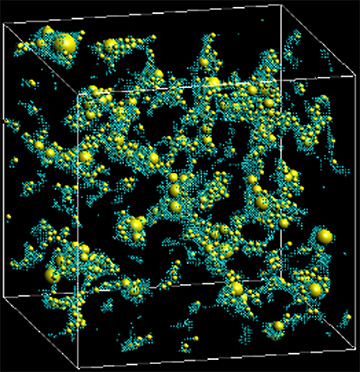
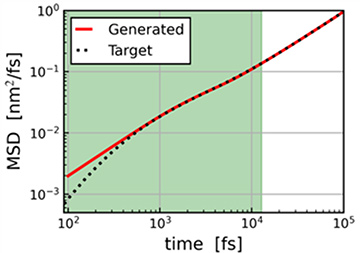
データサイエンスとの連携
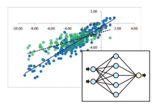
データサイエンスとの連携

ライフサイエンスのためのシミュレーション
J-OCTAは、マテリアルサイエンスで培った技術を活かし、ライフサイエンス分野にも強力な機能を提供しています。製剤設計では、DDS(ドラッグデリバリーシステム)向けの脂質ナノ粒子(LNP)、固体分散体、薬剤の溶解性の評価などに対応可能です。バイオマテリアル分野では、セルロースなどの糖鎖や脂質の分子構造に基づく物性予測、生体適合性や界面活性の評価に貢献します。創薬分野では、タンパク質とリガンドの結合構造探索や親和性評価を高精度に実施できます。J-OCTAはAI技術との連携も進めており、構造予測からシミュレーションまでを一貫して支援するプラットフォームとして進化を続けています。
全原子モデリング
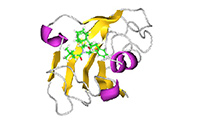
全原子モデリング
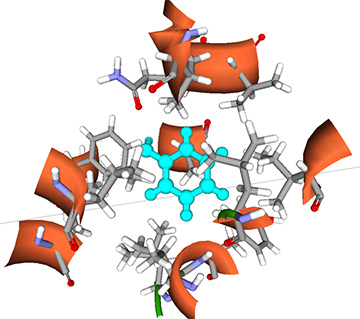
粗視化モデリング
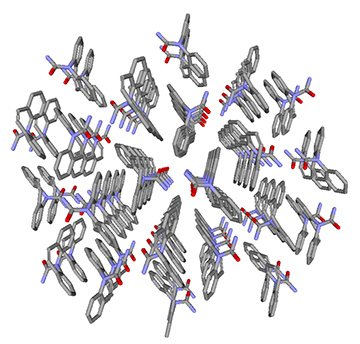
粗視化モデリング


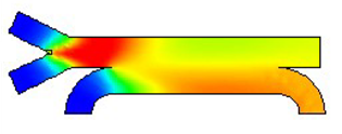
マイクロ流体・プロセスシミュレーション

マイクロ流体・プロセスシミュレーション
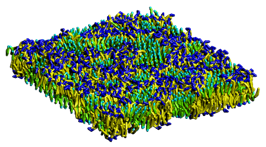
豊富なシミュレーションエンジン群
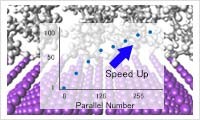
シミュレーション結果の解析

マテリアルズ・インフォマティクス
マテリアルズ・インフォマティクスの実現に必要なデータサイエンス機能を集めて、MI-Suiteというパッケージとしてご提供しています。J-OCTAのシミュレーション技術をデータサイエンス技術とつなぐことで、データドリブン型の材料設計をサポートします。
MI-Suite

MI-Suite
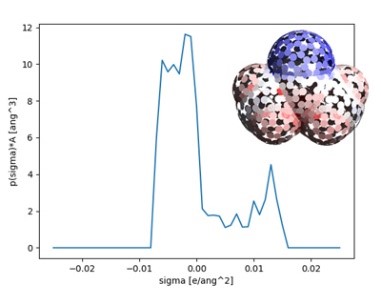
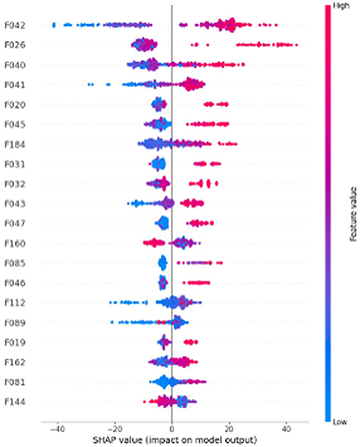
事例データベース・サポート・コンサルティングサービス
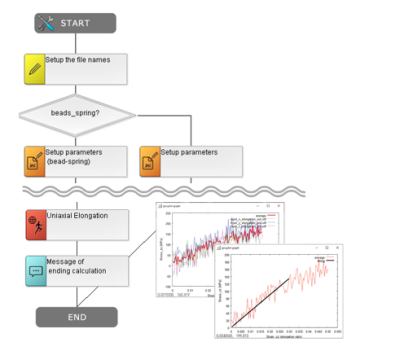
多くの場合、分子シミュレーションはマルチスケール&マルチフィジクスで高度な知識が必要となり、また実際にはどうなっているか分からないミクロな構造を仮説を立てて作ることが多く、初心者には難しさもありますが、J-OCTAの事例データベース(DB)やチュートリアル、シナリオ機能を活用することで着実に取り組めます。シナリオ機能では操作手順をフローチャート化して再利用でき、誤操作の回避やチーム内共有、大量な入力ファイルのバッチ処理にも便利です。
豊富な事例集とシナリオ機能

豊富な事例集とシナリオ機能
サポートとコンサルティングサービス

- ※ J-OCTAは産学連携プロジェクトで開発された ソフトマテリアルに対する統合的なシミュレータ「OCTA」の商用版ソフトウェアです。OCTAはオープンソースソフトウェアです。
- ※ SIESTAはスペイン等の研究者によって開発された密度汎関数法(DFT)のソフトウェアです。SIESTAはスペインSIMUNE社の登録商標です。
- ※ GENESISは理化学研究所を中心に開発している分子動力学ソフトウェアで、フリーソフトウェア(LGPLv3)として配布されています。
[1] C. Kobayashi, J. Jung, Y. Matsunaga, T. Mori, T. Ando, K. Tamura, M. Kamiya, and Y. Sugita, J. Compute. Chem. 38, 2193-2206 (2017).
[http://dx.doi.org/10.1002/jcc.24874]
[2] J. Jung, T. Mori, C. Kobayashi, Y. Matsunaga, T. Yoda, M. Feig, and Y. Sugita, WIREs Comput. Mol. Sci., 5, 310-323 (2015).
[http://dx.doi.org/10.1002/wcms.1220]
[3] J.Jung, K.Yagi, C.Tan, H.Oshima, T.Mori, I.Yu, Y.Matsunaga, C.Kobayashi, S.Ito, D.Ugarte La Torre, Y.Sugita, J. Phys. Chem. B 128, 25, 6028-6048 (2024).
[https://doi.org/10.1021/acs.jpcb.4c02096]
[4] ラグメント分子軌道計算エンジンABINIT-MP の文献:S. Tanaka, Y. Mochizuki, Y. Komeiji, Y. Okiyama, K. Fukuzawa, Phys. Chem. Chem. Phys. 16 (2014) 10310-10344 [https://doi.org/10.1039/C4CP00316K]
[5] FCEWS(FMO-DPD)の文献:K. Okuwaki, Y. Mochizuki, H. Doi, T. Ozawa, J. Phys. Chem. B, 122 (2018) 338-347[https://doi.org/10.1021/acs.jpcb.7b08461]
関連情報
お役立ち資料

ソリューション





セミナー・イベント






技術ブログ

