
Multiscale Modeling and Simulation Platform for Materials and Life Sciences
J-OCTA
Multiscale Modeling and Simulation Platform for Materials and Life Sciences
J-OCTA
Calculation of Li-Ion Battery Electrolyte
Full-Atomistic Molecular Dynamics (FAMD) was used to evaluate the relationship between molecular structure and properties of battery electrolytes. Representative molecules (EC, DMC, PF₆) were modeled, and self-diffusion coefficients of Li⁺ ions in the bulk state and the effects of composition ratios were analyzed.
Use Cases Highlights
- Evaluation of molecular structure and properties of electrolytes
- Evaluation of Li⁺ ion diffusion coefficients
- Analysis of the effect of composition ratio
Molecular structure modeling
Representative molecular structures of EC, DMC, and PF₆ are modeled in J-OCTA. The COGNAC modeler allows setting and adjustment of force field parameters including point charges, with electrolyte-specific parameters incorporated.
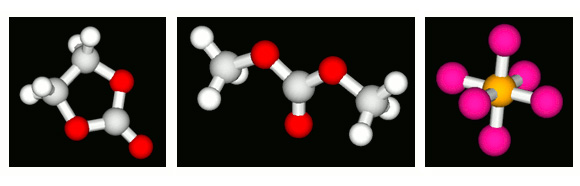
Molecular structure used in the calculation
Diffusion evaluation in bulk state
In the bulk state, the behavior of Li⁺ ions (shown as yellow particles) was analyzed using long-time relaxation calculations with Molecular Dynamics. The influence of self-diffusion coefficients and composition ratios of electrolyte components on diffusion can be evaluated.
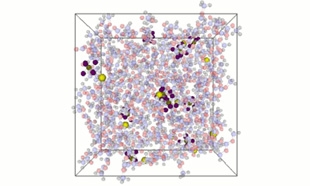
Bulk-state structure
Inquiries Regarding Products
Have questions about product implementation? Contact us today.
Related Information
Solutions

Materials Simulation and Materials Informatics (Automotive)
Materials development through the integration of simulation and data science (Materials DX)
Materials Simulation and Materials Informatics (Mobility)
Materials development through the integration of simulation and data science (Materials DX)

Materials Simulation and Materials Informatics (Electrical Equipment/Precision Equipment/Semiconductors/Heavy Electrical Equipment)
Materials development through the integration of simulation and data science (Materials DX)

