J-OCTA
J-OCTA
Outstanding Features and Services of J-OCTA
J-OCTA is a multiscale simulation software platform that supports a wide range of applications—from materials science fields such as polymer materials, composite materials, energy materials, electronics, and functional thin films/coatings, to life science fields such as drug discovery and formulation, and biomaterials that bridge both domains. It predicts structures and properties from the atomistic/molecular scale to the micrometer scale, helping to understand complex phenomena and generate new design guidelines. Simulators can be linked on a common platform, and integration with machine learning and data science for materials informatics is also available. This enables greater accuracy and efficiency in research and development, accelerating innovation in both materials science and life sciences. With extensive adoption and robust support, J-OCTA can be used with confidence by both first-time users and experienced researchers.
- Simulation for Materials Science
- Simulation for Life Sciences
- Materials Informatics
- Support and Consulting Services
Simulation for Materials Science
In materials design, understanding and controlling multiscale structures—from the nano to the macro level—is essential. J-OCTA is an integrated simulation platform that spans from quantum theory to continuum theory, corresponding to each scale. It employs full-atomistic models, coarse-grained models, and continuum models to support everything from predicting structures and properties to process design. It enables evaluation of a wide range of physical properties, including mechanical, thermal, electrical, and optical characteristics, and allows multiscale coupling as well as integration with data science. With a wealth of case studies and tutorials, even first-time users can confidently get started, while researchers and engineers can deepen their understanding of structures and behaviors and apply these insights to design and development.
Full-Atomistic Modeling
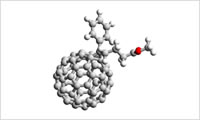
Full-Atomistic Modeling

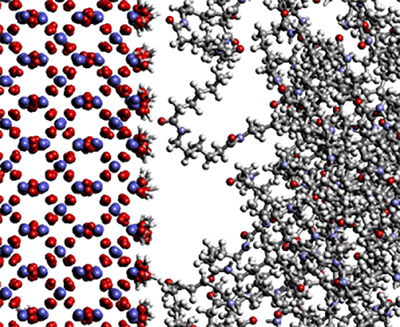
Coarse-Grained Modeling

Coarse-Grained Modeling


Micromechanics, Microscale Fluid Dynamics, and Process Simulation

Micromechanics, Microscale Fluid Dynamics, and Process Simulation
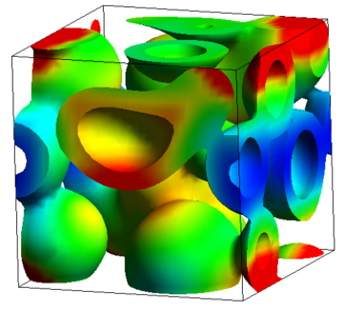
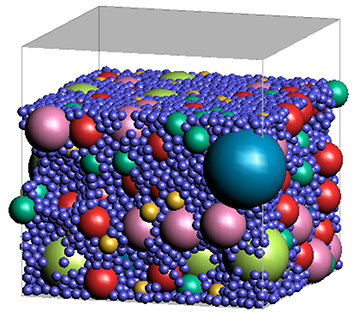
- Simulation of phase separation process in polymeric membranes
- Calculation of pressure and porosity in the forming process (calendaring) of battery electrodes
- Simulation of evaporation of liquid film
- Thermal conductivity calculations for filler resin composites
- Nonlinear Mechanical Properties of Composites (Linked with Ansys LS-DYNA)
Wide Range of Simulation Engines
Wide Range of Simulation Engines
Analysis of Simulation Results and Property Estimation

Analysis of Simulation Results and Property Estimation

Data Science Integration
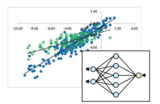
Data Science Integration

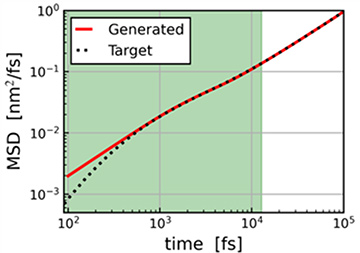
Simulation for Life Science
J-OCTA leverages the technologies cultivated in materials science to deliver powerful capabilities for the life sciences as well. In pharmaceutical formulation design, it supports the evaluation of lipid nanoparticles (LNP) for drug delivery systems (DDS), solid dispersions, and drug solubility. In biomaterials, it enables property prediction based on the molecular structures of polysaccharides such as cellulose and of lipids, along with the evaluation of biocompatibility and interfacial activity. In drug discovery, it enables high-accuracy exploration of protein–ligand binding structures and evaluation of binding affinity. J-OCTA is also advancing integration with AI technologies, continuing to evolve as a platform that supports the entire workflow from structure prediction to simulation.
Full-Atomistic Modeling
Full-Atomistic Modeling
Coarse-Grained Modeling
Coarse-Grained Modeling


Microscale Fluid Dynamics and Process Simulation

Microscale Fluid Dynamics and Process Simulation
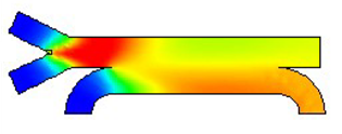
Wide Range of Simulation Engines
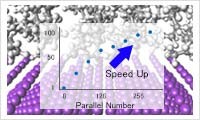
Analysis of Simulation Results

Materials Informatics
We offer the data science capabilities necessary for materials informatics in a dedicated software package called MI-Suite. By connecting J-OCTA’s simulation technologies with data science techniques, it supports data-driven materials design.
MI-Suite

MI-Suite
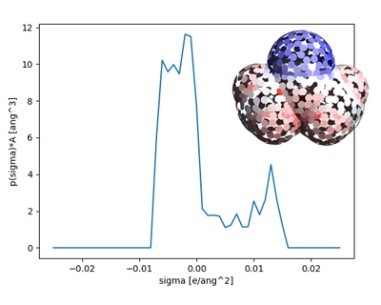
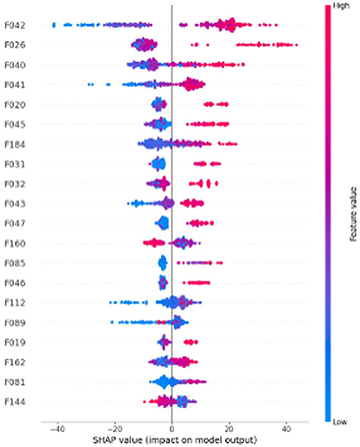
Case Study Database, Support, and Consulting Services
In many cases, molecular simulations require advanced knowledge in multi-scale and multi-physics approaches, and often involve building hypothetical microscopic structures whose actual states are unknown. This can make the work challenging for beginners. However, by utilizing J-OCTA’s case study database (DB), tutorials, and scenario functions, users can proceed with confidence. The scenario function allows operation procedures to be visualized as flowcharts for reuse, helping to prevent operation errors, facilitate team sharing, and enable batch processing of large numbers of input files.
Extensive Case Study Collection and Scenario Function

Extensive Case Study Collection and Scenario Function
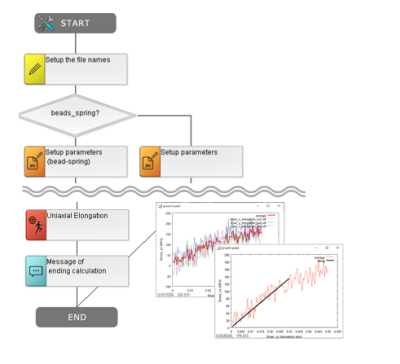
Support and Consulting Services

- ※
J-OCTA is the commercial version of the integrated simulator for soft materials, called "OCTA," that was developed through an industry-university cooperative project. OCTA is open-source software.
See more details for OCTA >> [https://octa.jp/] - ※
SIESTA is open source Density Functional Theory (DFT) software developed by a group including Universities mainly in Spain. SIESTA is a registered trade-mark of SIMUNE.
SIMUNE Website >> [https://www.simuneatomistics.com/]
SIESTA Open Source Software Website >> [https://siesta-project.org/siesta/] - ※
GENESIS is molecular dynamics software developed mainly by RIKEN and distributed as free software (LGPLv3).
GENESIS Website >>[https://mdgenesis.org/]
Reference:
[1] C. Kobayashi, J. Jung, Y. Matsunaga, T. Mori, T. Ando, K. Tamura, M. Kamiya, and Y. Sugita, J. Compute. Chem. 38, 2193-2206 (2017).
[http://dx.doi.org/10.1002/jcc.24874]
[2] J. Jung, T. Mori, C. Kobayashi, Y. Matsunaga, T. Yoda, M. Feig, and Y. Sugita, WIREs Comput. Mol. Sci., 5, 310-323 (2015).
[http://dx.doi.org/10.1002/wcms.1220]
[3] J.Jung, K.Yagi, C.Tan, H.Oshima, T.Mori, I.Yu, Y.Matsunaga, C.Kobayashi, S.Ito, D.Ugarte La Torre, Y.Sugita, J. Phys. Chem. B 128, 25, 6028-6048 (2024).
[https://doi.org/10.1021/acs.jpcb.4c02096] - ※
References for the fragment molecular orbital calculation engine ABINIT-MP:
S. Tanaka, Y. Mochizuki, Y. Komeiji, Y. Okiyama, K. Fukuzawa, Phys. Chem. Chem. Phys. 16 (2014) 10310-10344 [https://doi.org/10.1039/C4CP00316K]
References for FCEWS (FMO-DPD):
K. Okuwaki, Y. Mochizuki, H. Doi, T. Ozawa, J. Phys. Chem. B, 122 (2018) 338-347[https://doi.org/10.1021/acs.jpcb.7b08461]
Related Information
Solutions