
Multiscale Modeling and Simulation Platform for Materials and Life Sciences
J-OCTA
Multiscale Modeling and Simulation Platform for Materials and Life Sciences
J-OCTA
DFT-MD study for interfacial systems
The SIESTA interfacial energy calculation tool in J-OCTA was used to analyze interactions between alkane molecules adsorbed on a copper (111) surface. Adsorption energies obtained by density functional theory (DFT), a first-principles calculation, were fitted with a generalized Lennard-Jones function, and the obtained parameters were used in Full-Atomistic Molecular Dynamics (FAMD) calculations. Changes in adsorption energy with increasing carbon number were also reproduced.
Use Cases Highlights
- Application example of J-OCTA SIESTA interfacial energy calculation tool
- Determination of parameters for MD simulations
- Applicable to various phenomena at inorganic–organic interfaces
Application example of J-OCTA SIESTA interfacial energy calculation tool
An example of alkane molecules adsorbed on a copper (111) surface is shown. The J-OCTA SIESTA Interfacial Energy Tool is used to determine force field parameters for organic–inorganic interfaces, and Full-Atomistic Molecular Dynamics (FAMD) simulations are performed.
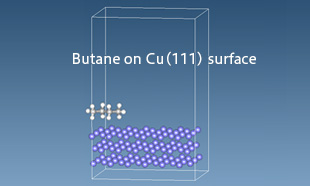
Alkane molecules adsorbed on Cu(111) surface
Determination of parameters for MD simulations
The interaction energies between alkane molecules (propane, butane, pentane, hexane) and a copper surface were calculated using DFT with SIESTA, and the force field parameters used in Molecular Dynamics (MD) were fitted with a Generalized Lennard-Jones function.
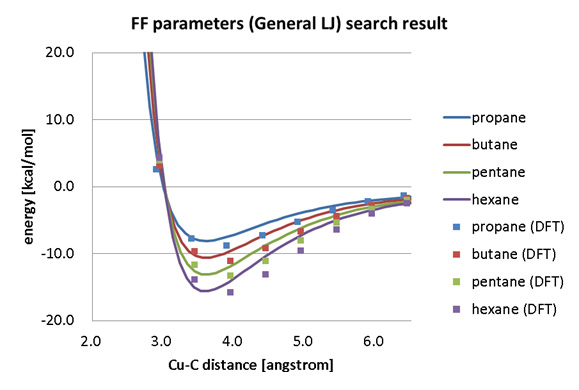
Force field parameter fitting results
Details of analysis
Related information
Inquiries Regarding Products
Have questions about product implementation? Contact us today.
Related Information
Solutions

Materials Simulation and Materials Informatics (Automotive)
Materials development through the integration of simulation and data science (Materials DX)
Materials Simulation and Materials Informatics (Mobility)
Materials development through the integration of simulation and data science (Materials DX)

Materials Simulation and Materials Informatics (Electrical Equipment/Precision Equipment/Semiconductors/Heavy Electrical Equipment)
Materials development through the integration of simulation and data science (Materials DX)

