
Designing Plastic Recycling with Molecular Simulation
Plastic recycling includes a range of approaches, mechanical, dissolution (solvent-based), chemical, and biological (enzymatic) routes, as well as strategies enabled by new materials such as vitrimers. By combining molecular simulation with machine learning, it is possible to examine in advance “hard-to-observe factors” such as interfacial structure, phase separation, solvent selection, and rate-limiting steps in reactions, which can improve the efficiency of experimental work. This article provides a practical, introductory overview of these topics.
1.Introduction
Plastics are essential in daily life, and the importance of recycling continues to grow as sustainability becomes a higher priority. At the same time, “recycling” does not refer to a single method. Examples include:
- Mechanical recycling: physically shredding used plastics and remolding them.
- Dissolution recycling: selectively extracting and purifying a target resin using solvents.
- Chemical recycling: decomposing polymers via chemical reactions or heat back into monomers.
- Biological (enzymatic) recycling: promoting decomposition under mild conditions using enzymes.
- Recycling approaches enabled by new materials such as vitrimers, which can be reshaped upon heating.[1]
Because the options are diverse, it is not always straightforward to determine which method, under which conditions, is best suited for a given material. In this article, molecular simulation and data-science techniques are introduced from the perspective of their applicability across different recycling methods. By examining molecular-scale phenomena in advance, such as the state of polymer–polymer interfaces, solubility in solvents, or bottlenecks in chemical reactions, it becomes possible to reduce trial-and-error in experiments and accelerate development.
2.Overview of Simulation Methods
Molecular simulation covers multiple methods. By selecting approaches appropriate to the length/time scale of interest, one can connect microscopic molecular behavior to macroscopic material properties. For more details, see Ref.[2]
- Quantum chemical calculations (e.g., DFT, QM/MM)
- These methods capture chemical reactions. They can evaluate the height of the “energy barrier” a reaction must overcome (activation energy) and compare plausible reaction pathways.[5, 7]
- Molecular dynamics (MD)
- MD tracks the motion of atoms and molecules over time. It is suitable for observing behaviors such as diffusion, or how polymer chains at interfaces contribute during deformation.[2]
- Coarse-grained / mesoscale simulation (e.g., CGMD, DPD)
- Instead of tracking individual atoms, groups of atoms are represented as coarse-grained segments. This enables efficient prediction of larger-scale structures (tens to hundreds of nanometers). These approaches are effective for understanding phase separation between different polymers and the resulting interfacial morphologies.[2]
- Continuum mechanics (CAE/FEM)
- Materials are treated as continuous media. This allows evaluation of how predicted microstructures affect macroscopic mechanical properties such as strength, stiffness, and deformability, linking microscopic morphology to macroscopic behavior.[2]
- Thermodynamics / solubility prediction (e.g., QSPR, COSMO-RS/SAC)
- Using machine-learning models trained on experimental data (QSPR) or physics-based models based on molecular surface charge characteristics (COSMO methods), thermodynamic properties such as miscibility and solubility can be predicted rapidly. This enables efficient screening of promising solvents from a large candidate space before experiments are performed.
3. Mechanical Recycling
Mechanical recycling typically involves washing, shredding, melting, and reprocessing waste plastics into new products. Because it is a physical reuse route, it can be cost-effective and relatively quick to implement; however, property degradation and contamination/mixing are major challenges.
When different resins are mixed, phase-separated structures often form because polymers do not fully mix at the molecular level. In addition, degradation during use and recycling can change molecular weight (chain scission) and may also generate crosslinked structures. Predicting all of these effects simultaneously with molecular simulation is difficult. Still, under practical assumptions, simulation can be applied to topics such as the following:
- Interfacial tension
- Predict interfacial structure between different resins and evaluate interfacial tension. Interfaces are not always sharp; an intermixing region may exist. When the interfacial width is typically a few nanometers (and may reach ~10 nm depending on conditions), coarse-grained/mesoscale models are applicable.
- Effects of compatibilizers
- Evaluate how compatibilizers hold different resins together at the interface. This includes assessing what molecular architectures are effective and how much compatibilizer is needed. When the interfacial width is on the order of ~10 nm, coarse-grained models are applicable.
- Impact of crosslinked structures
- Assuming crosslinks are formed by chemical reactions, evaluate how crosslinking affects mechanical properties. Coarse-grained models are applicable.
- Viscoelasticity
- Assuming a molecular weight distribution and/or formation of crosslinks, evaluate how these factors affect viscoelastic properties. Coarse-grained models are applicable.
- Microstructure and properties
- Use coarse-grained simulation to analyze mechanisms of phase-separated structure formation (e.g., sea–island morphologies). Evaluate how molecular structure and formulation ratios affect morphology, and assess relationships to mechanical properties such as average modulus. Mesoscale models are applicable.
Because processing steps such as kneading and injection molding also influence the final material structure, simulations that incorporate flow history can be important for more realistic predictions.[3]
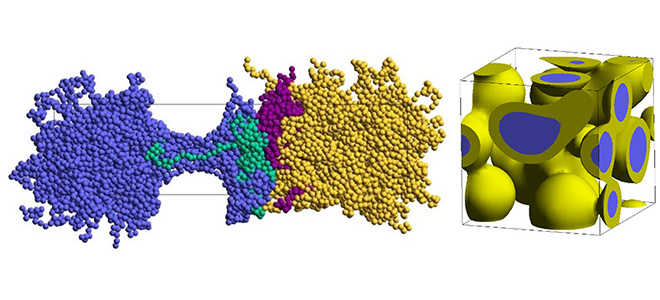
Figure 1. Interfaces formed between different resins and a compatibilizer (left); phase-separated morphology (right).
4.Dissolution Recycling
In processes that selectively dissolve and recover a specific resin from multilayer films or composites, identifying an appropriate solvent is essential. However, the number of potential solvents is enormous, and testing each one experimentally is not realistic. Molecular simulation and data science can support solvent selection in workflows such as the following:
- QSPR (Quantitative Structure–Property Relationship)
- A machine-learning model is trained on large experimental datasets of polymer–solvent solubility and used to predict solubility from molecular structural information. This can generate a short list of promising solvent candidates in relatively short time.
- COSMO-RS/SAC
- Based on quantum chemical calculations, electrostatic features of each molecule (σ-profiles) are computed, and thermodynamic stability upon mixing (e.g., activity coefficients) is predicted using a physics-based model. This helps identify combinations with higher miscibility and selectivity among candidates screened by QSPR.
5. Chemical Recycling
Chemical recycling aims to decompose polymers back into monomers. It is influenced by many factors, e.g. catalysts, temperature, solvents, and crystallinity of the feedstock plastic, making it a complex process. To improve efficiency, it is important first to identify the bottleneck that determines the overall speed, i.e., the rate-limiting step.
For example, ester bonds in PET (polyethylene terephthalate) are relatively easier to break down, whereas designing reaction pathways that efficiently break the strong carbon–carbon backbone in polyolefins such as PE (polyethylene) and PP (polypropylene) remains a challenge.[4, 9] Molecular simulation can contribute both to bottleneck identification and to exploration of new reaction routes.
- Evaluation of activation energy
- For a chemical reaction to proceed, it must overcome an “energy barrier.” Quantum chemical calculations can compute the barrier height (activation energy) along reaction pathways and evaluate which step is the most energetically difficult (and therefore slowest) using model structures. While quantum chemical methods are commonly used, recent studies also report attempts using full-atomistic (all-atom) MD with machine-learning force fields.[5]
- Evaluation of accessibility to catalysts
- Even a strong catalyst is ineffective if the target polymer chains cannot approach its active sites. MD can evaluate polymer swelling in solvent and accessibility around catalysts, helping assess whether diffusion may be rate-limiting.
![Figure 2. Thermal decomposition process of polystyrene (PS) studied using MD and a machine-learning force field [5]. (Used under the CC BY Attribution 4.0 International license. J-OCTA was not used in the original study.)](https://www.jsol-cae.com/tech-blog/aluome00000000ej-img/002.jpg)
Figure 2. Thermal decomposition process of polystyrene (PS) studied using MD and a machine-learning force field[5].
(Used under the CC BY Attribution 4.0 International license. J-OCTA was not used in the original study.)
6. Biological (Enzymatic) Recycling
In recent years, discoveries of enzymes that can decompose specific plastics have increased interest in biological recycling. Enzymes act as highly specific “molecular scissors” that break targeted chemical bonds. For PET degradation, it is known that combining PETase (which first decomposes PET) with MHETase (which further decomposes the degradation products) can improve decomposition rate and yield.[6] Molecular simulation can identify reaction bottlenecks at the molecular level.[7] Such information can also be used when considering experimental conditions such as temperature and pH.
- Optimization of the active site
- By substituting amino acids in the active site (the enzyme’s “blade”), QM/MM calculations (quantum chemistry for the region where reaction detail is required, and molecular mechanics for the remainder) can evaluate how performance may change. This can support the design of modified enzymes that may lower reaction energy barriers.
- Design of substrate pretreatment
- For enzymes to work efficiently, they must access the plastic substrate. Pretreatments such as increasing surface area by grinding or increasing the amorphous fraction may influence reactivity. Simulation can evaluate molecular-level trends to guide experimental planning.
![Figure 3. PET degradation by two enzymes from Ideonella sakaiensis [6][7]. Coordinated action of PETase and MHETase induces the reaction PET -> MHET -> terephthalic acid and ethylene glycol. (Partially modified and used under the CC BY Attribution 4.0 International license. J-OCTA was not used in the original study.)](https://www.jsol-cae.com/tech-blog/aluome00000000ej-img/003.jpg)
Figure 3. PET degradation by two enzymes from Ideonella sakaiensis[6][7]. Coordinated action of PETase and MHETase induces the reaction PET -> MHET -> terephthalic acid and ethylene glycol.
(Partially modified and used under the CC BY Attribution 4.0 International license. J-OCTA was not used in the original study.)
7. Recycling Enabled by New Materials such as Vitrimers
Conventional thermosetting resins have a limitation: once cured, they cannot be reshaped, which makes recycling difficult. Vitrimers have attracted attention as materials that address this issue. Vitrimers contain “dynamic covalent bonds” that can reversibly rearrange within the network upon heating. This enables the material to flow and be remolded while maintaining a crosslinked structure. Materials whose covalent network connectivity changes slowly over time are collectively referred to as Covalent Adaptable Networks (CANs), and vitrimers are a representative example.
A commonly used indicator for organizing this behavior is the topology freezing transition temperature (Tv). Tv refers to the temperature at which bond-exchange reactions effectively stop (i.e., the network topology becomes frozen), and it is typically evaluated experimentally (e.g., via rheology). When heated above Tv, bond exchange becomes active and the viscosity can decrease substantially, improving processability.[8]
Using MD simulations (full-atomistic (all-atom) and/or coarse-grained), it is possible to examine temperature-dependent behavior such as molecular/network mobility and the extent of bond rearrangement. With simulation, one can roughly grasp relationships such as temperature versus molecular mobility before experiments, which can provide guidance for material selection and design suitable for targeted processing conditions.[8]
![Figure 4. Types of CANs (Covalent Adaptable Networks) [8]. CANs are classified into dissociative and associative systems; vitrimers are a representative associative CAN. (Used under the CC BY Attribution 4.0 International license. J-OCTA was not used in the original study.)](https://www.jsol-cae.com/tech-blog/aluome00000000ej-img/004.jpg)
Figure 4. Types of CANs (Covalent Adaptable Networks)[8]. CANs are classified into dissociative and associative systems; vitrimers are a representative associative CAN.
(Used under the CC BY Attribution 4.0 International license. J-OCTA was not used in the original study.)
8. Closing Remarks
The multi-scale simulations introduced here—from quantum chemistry to continuum mechanics—and the narrowing of conditions through combination with data science require careful integration across methods. Such integrated workflows can be supported by an integrated environment (for example, a platform such as “J-OCTA”).
- van den Tempel, P., Picchioni, F., Recycling 2025, 10, 1.
(https://doi.org/10.3390/recycling10010001) - https://www.jsol-cae.com/en/tech-blog/20220501.html
- Deng,L., et al., Polymers, 2021,13, 3783.
(https://doi.org/10.3390/polym13213783) - Clark, R. A., Shaver, M. P., Chem. Rev. 2024, 124, 2617–2650.
(https://doi.org/10.1021/acs.chemrev.3c00739) - Mieda, S., ACS Omega 2025, 10, 5973–5980.
(https://doi.org/10.1021/acsomega.4c09953) - Knott, B. C., et al. PNAS 2020, 117, 25476–25485.
(https://doi.org/10.1073/pnas.2006753117) - dos Santos, A. M., et al., J. Phys. Chem. B 2024, 128, 7486–7499.
(https://doi.org/10.1021/acs.jpcb.4c02207) - Karatrantos, A. V., et al., Polymers 2024, 16, 1373.
(https://doi.org/10.3390/polym16101373) - Peti, D., et al., Polymers 2025, 17, 603.
(https://doi.org/10.3390/polym17050603)
Related Information
Categories
New articles
- Simulation of Coating-Film Drying : A Practical Introduction to 1D/3D Analysis for Coating, Painting, and Electrode Processes
- Designing Plastic Recycling with Molecular Simulation
- What is Materials Informatics (MI)?
Next-generation materials development accelerated by AI × simulation
- Electrical conduction as a multiscale simulation
- Development of force fields used in molecular dynamics calculation
- What you need to know when using molecular modeling and simulation for materials design
- Materials & Process Informatics
- Machine Learning for Materials Design in J-OCTA - - Simulation of lithium-ion batteries
- Drug Discovery and Formulation
- Simulation of Polymeric Materials : Overview and Examples
