

第一原理電子状態計算ソフトウェア
ASAP
第一原理電子状態計算ソフトウェア
ASAP
分子間相互作用の計算
本事例では、SIESTAを用いてS22ベンチマークセットに含まれる22の分子性錯体の相互作用エネルギーを計算した。DRSLL、LMKLL、KBM、C09、BH、VVなどのVan der Waals汎関数とGrimmeの分散力補正を用いたPBE-D汎関数を比較し、Counterpoise補正を施した結果をCCSD(T)/CBS CPの参照値と照合。絶対誤差の標準偏差(SDEV)と平均二乗誤差(MAE)により評価した。
解析・利用例のポイント
- 分子会合体の解析に適用可能
- 相互作用エネルギーの比較
- 誤差評価のカテゴリ別傾向
分子会合体の構造一覧
S22ベンチマークに含まれる22の分子性錯体の構造を示す図。水素結合、分散力、双極子相互作用など多様な相互作用を含む構造が視覚的に確認できる。
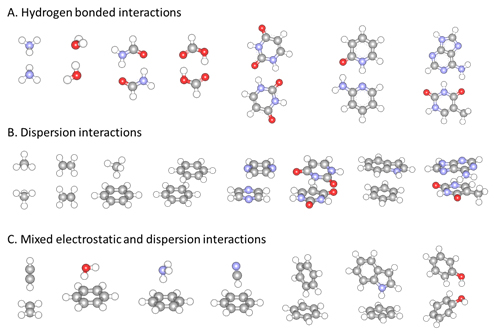
S22ベンチマーク構造一覧
相互作用エネルギーの比較
各汎関数による相互作用エネルギーと参照値との誤差を示す図。SDEVとMAEにより、汎関数ごとの精度が比較されている。
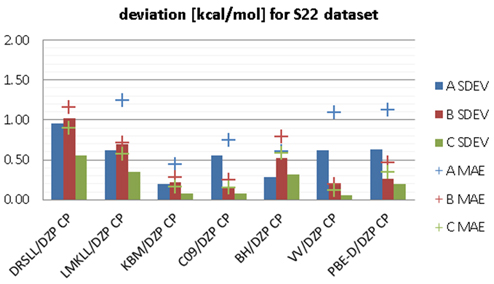
相互作用エネルギーの誤差比較
誤差評価のカテゴリ別傾向
A(水素結合)、B(分散力)、C(双極子相互作用)カテゴリごとの誤差傾向を示す図。汎関数の特性により得意・不得意な相互作用があることが確認できる。

相互作用カテゴリ別誤差傾向
解析内容の詳細
関連情報
セミナー・イベント

ソリューション

材料シミュレーションとマテリアルズ・インフォマティクス(自動車)
データとシミュレーションを連携した材料開発(マテリアルDX)
材料シミュレーションとマテリアルズ・インフォマティクス(輸送機器)
データとシミュレーションを連携した材料開発(マテリアルDX)

材料シミュレーションとマテリアルズ・インフォマティクス(電気/精密機器/半導体/重電)
シミュレーションとデータサイエンスの連携による材料開発(マテリアルDX)

材料シミュレーションとマテリアルズ・インフォマティクス(化学/材料)
シミュレーションとデータサイエンスの連携による材料開発(マテリアルDX)

シミュレーションとデータサイエンスの連携による医薬品開発(医薬品)
創薬と製剤分野への先端シミュレーション技術とAIの適用

リチウムイオン電池の材料シミュレーションとマテリアルズ・インフォマティクス
シミュレーションとデータサイエンスの連携による電池材料開発(マテリアルDX)
