

第一原理電子状態計算ソフトウェア
ASAP
第一原理電子状態計算ソフトウェア
ASAP
顔料の表面エネルギー
本事例では、シアン系有機顔料である銅フタロシアニン(CuPc)の001面および011面の表面エネルギーを、密度汎関数法(DFT)に基づくSIESTAを用いて計算した。バルクモデルと表面モデルのエネルギー差からγを算出し、001面は0.12 J/m²、011面は0.21 J/m²と評価された。分子間相互作用の影響が011面で強く現れる傾向が確認された。
解析・利用例のポイント
- CuPc顔料の表面エネルギー評価
CuPcの表面構造
銅フタロシアニンの001面と011面の表面構造を示す図。分子性結晶としての特徴を持ち、面方位による分子間相互作用の違いが視覚的に確認できる。
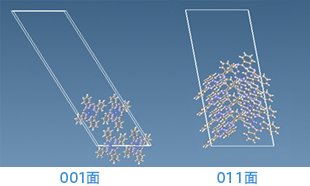
CuPcの001面・011面構造
解析内容の詳細
関連情報
セミナー・イベント

ソリューション

材料シミュレーションとマテリアルズ・インフォマティクス(自動車)
データとシミュレーションを連携した材料開発(マテリアルDX)
材料シミュレーションとマテリアルズ・インフォマティクス(輸送機器)
データとシミュレーションを連携した材料開発(マテリアルDX)

材料シミュレーションとマテリアルズ・インフォマティクス(電気/精密機器/半導体/重電)
シミュレーションとデータサイエンスの連携による材料開発(マテリアルDX)

材料シミュレーションとマテリアルズ・インフォマティクス(化学/材料)
シミュレーションとデータサイエンスの連携による材料開発(マテリアルDX)

シミュレーションとデータサイエンスの連携による医薬品開発(医薬品)
創薬と製剤分野への先端シミュレーション技術とAIの適用

リチウムイオン電池の材料シミュレーションとマテリアルズ・インフォマティクス
シミュレーションとデータサイエンスの連携による電池材料開発(マテリアルDX)
