
分子シミュレーションで設計するプラスチックリサイクル
プラスチックリサイクルには、機械・溶解・化学・生物分解といった手法に加え、ビトリマーなど新素材を活用したアプローチがあります。分子シミュレーションと機械学習の活用により、界面や相分離、溶媒選定、反応の律速など“見えない要因”を事前に可視化し、実験の効率を高めることができます。本稿では、これらのトピックについて実務的なポイントをやさしく概観します。
1. はじめに
私たちの生活に欠かせないプラスチック。その持続可能性を高めるため、リサイクルの重要性はますます高まっています。一口にリサイクルと言っても、その方法は多様です。
使用済みプラスチックを物理的に粉砕し、再び成形する「機械リサイクル」、特定の樹脂だけを溶媒で選び出して精製する「溶解リサイクル」、ポリマーを化学反応や熱によって原料のモノマーにまで分解する「化学リサイクル」、酵素の力を借りて穏やかな条件で分解を進める「生物(酵素)リサイクル」、熱を加えることで再成形が可能になる「ビトリマー」など「新素材を活用したリサイクル」、さまざまな方法が開発されています [1]。
しかし、選択肢が多岐にわたるということは、裏を返せば「どの材料に、どの方法を、どの条件で適用するのが最適か」という問いに答えるのが難しいということでもあります。この記事では、分子シミュレーションやデータサイエンス技術について、各種プラスチックリサイクル方法への適用可能性という視点からご紹介します。分子レベルの現象、たとえば樹脂が混ざり合う界面の状態、溶媒への溶けやすさ、化学反応のボトルネックなどを事前に「見える化」することで、実験の回数を減らし、開発を加速させることができます。
2. シミュレーション手法の紹介
分子シミュレーションは、現象のスケールに応じて適切な手法を使い分けることで、ミクロな分子の挙動からマクロな材料特性までを予測します。詳細は文献[2] などを参照してください。
- 量子化学計算(DFT、QM/MMなど)
- 化学反応を捉えるための手法です。反応が進むために乗り越えなければならないエネルギーの山(活性化エネルギー)の高さや、効率的な反応経路を評価できます[5, 7]。
- 分子動力学(MD)
- 原子や分子の動きを時々刻々と追いかける手法です。分子がどのように拡散していくか、界面でのポリマー鎖が変形時にどのような役割を担うか、といった振る舞いを観察するのに適しています[2]。
- 粗視化/メソスケールシミュレーション(CGMD、DPDなど)
- 個々の原子を追うのではなく、分子のグループを1つの粒子とみなす(粗視化する)ことで、より大きなスケール(数十〜数百ナノメートル)の構造を効率的に予測します。異なる種類のポリマーがどのように相分離し、どのような界面形態を形成するかといった、材料の微細構造を把握するのに威力を発揮します[2]。
- 連続体力学(CAE/FEM)
- 分子レベルの構造から離れ、材料を連続的な物体として扱う手法です。シミュレーションで予測された微細構造が、材料全体の強度や剛性、変形のしやすさといった機械的特性にどう影響するかを評価し、ミクロな形態とマクロな物性を結びつけます[2]。
- 熱力学・溶解性推算(QSPR、COSMO-RSなど)
- 実験データから学習したモデル(QSPR)や、分子の表面電荷に基づいた物理モデル(COSMO系)を用いて、物質の混和性や溶解度といった熱力学的な性質を高速に予測します。これにより、実験を行う前に膨大な数の候補がある溶媒の中から、有望なものを効率的に絞り込むこと(スクリーニング)が可能です。
3. 機械リサイクル
機械的(メカニカル)な手法では、廃プラスチックを洗浄、粉砕、溶融し、新たな製品に再加工します。物理的再利用のため、安価かつ即効性がありますが、性質の劣化や不純物混合の処理が課題となります。異なる種類の樹脂が混ざった状態ではしばしば分子レベルで完全に混ざり合わない相分離構造が形成されます。その他、劣化やリサイクルプロセスの中でポリマー鎖の切断により分子量(長さ)が変わること、架橋構造が形成されることも考えられます。分子シミュレーションでこれら全ての現象を予測することは困難です。リサイクル樹脂に特化した話ではないですが、さまざまな仮定をおきながら、以下のような課題に適用することが考えられます。
- 界面張力:
- 異なる樹脂間の界面構造を予測し界面張力を評価します。界面は明瞭でなく、2種の樹脂が混ざり合って存在する領域が存在することがあります。その幅が一般に数nm、条件により〜10nm程度になる場合、粗視化/メソスケールのモデルが適用されます。
- 相容化剤の効果:
- 異種樹脂の界面で両者をつなぎとめる相容化剤の効果を評価します。どのような分子構造の相容化剤が、どれくらいの量があれば最も効果的かを検討します。上述のように界面幅が10nm程度になる場合、粗視化モデルが適用されます。
- 架橋構造の影響:
- 化学反応により架橋が形成された場合を仮定して、機械特性への影響を評価します。粗視化モデルが適用されます。
- 粘弾性:
- 分子量分布や架橋構造の形成を仮定した場合の粘弾性特性への影響を評価します。粗視化モデルが適用されます。
- ミクロ構造と物性:
- 粗視化シミュレーションで相分離構造(海島構造など)の形成メカニズムを解析します。分子構造や配合比などが相分離構造に及ぼす影響、平均弾性率など機械特性との関係を評価します。メソスケールのモデルが適用されます。
成形時の練り込みや射出といった加工プロセスも最終的な材料構造に影響を与えるため、これらの流れの履歴を考慮したシミュレーションも、より現実に近い予測を行う上では重要となります[3]。
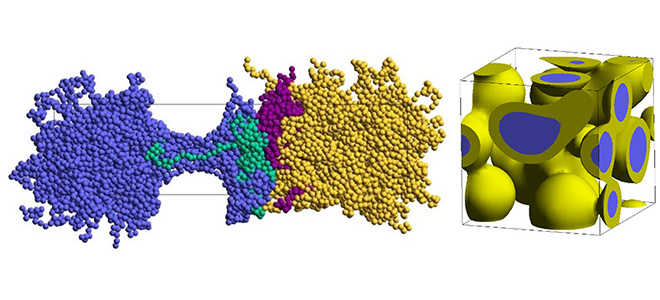
4. 溶解リサイクル
多層フィルムや複合材料から特定の樹脂だけを選択的に溶解・回収するプロセスでは、最適な溶媒を見つけ出すことが必要となります。しかし、候補となる溶媒は無数にあり、それらをひとつひとつ実験で試すのは非現実的です。そこで、以下のような分子シミュレーションとデータサイエンスが用いられます。
- QSPR(定量的構造物性相関):
- 既知の溶媒とポリマーの溶解性に関する膨大な実験データを機械学習させ、分子の構造情報から溶解度を予測するモデルを構築します。これにより、有望な溶媒候補をまとめた短いリストを比較的短時間で作成できます。
- COSMO-RS/SAC:
- 量子化学計算に基づいて各分子の静電的な特徴(σプロファイル)を算出し、それらを用いて混合時の熱力学的な安定性(活量係数など)を物理的に予測します。これにより、QSPRで絞り込んだ候補の中から、さらに混和性や選択性の高い組み合わせを見つけ出します。
5. 化学リサイクル
ポリマーを化学的にモノマーまで分解する化学リサイクルは、触媒、温度、溶媒、そして原料プラスチックの結晶性など、数多くの因子が複雑に絡み合うプロセスです。この複雑な反応系全体を効率化するには、まず全体のスピードを決定しているボトルネック、すなわち律速段階を特定することが重要です。たとえば、PET(ポリエチレンテレフタレート)のエステル結合は比較的分解しやすいですが、PE(ポリエチエン)やPP(ポリプロピレン)の強固な炭素-炭素主鎖を効率的に切断する反応経路の設計は、今なお大きな課題です[4, 9]。分子シミュレーションは、新たな反応ルートを探る上でも重要な役割を担います。
- 活性化エネルギーの評価:
- 化学反応が進むためには、ある種の「エネルギーの山」を越える必要があります。量子化学計算は、反応経路に沿ったこの山の高さ(活性化エネルギー)を精密に計算し、どのステップが最もエネルギー的に困難(=遅い)かを、モデル構造を用いて評価します。主に量子化学計算が用いられますが、最近では機械学習力場を用いた全原子分子動力学を用いる試みも報告されています[5]。
- 触媒へのアクセス性の評価:
- どんなに優れた触媒があっても、反応させたいポリマー鎖がその活性点に近づけなければ意味がありません。分子動力学法により、ポリマーが溶媒中でどれだけ膨潤するか、あるいは触媒の周りにどれだけアクセスしやすいかを評価し、拡散が律速になっている可能性を評価することができます。
![図2.MDと機械学習力場を用いたPSの熱分解プロセス [5] (CC BY Attribution 4.0 International ライセンスの下で使用)](/tech-blog/aluome00000000ej-img/002.jpg)
(CC BY Attribution 4.0 International ライセンスの下で使用)
6. 生物リサイクル
近年、特定のプラスチックを分解する酵素の発見が相次ぎ、生物リサイクルへの期待が高まっています。酵素は、特定の化学結合だけを狙って切断する、非常に精密な「分子のハサミ」です。たとえばPET分解では、まずPETを分解するPETaseと、その分解生成物をさらに細かく分解するMHETaseを組み合わせることで、分解速度と収率が向上することが知られています[6]。分子シミュレーションにより、どこが反応のボトルネックになっているかを分子レベルで特定できます[7]。このような情報は、温度や pH など実験条件の検討にも活用できます。
- 活性部位の最適化:
- 酵素の「刃」にあたる活性部位のアミノ酸を別のアミノ酸に置き換えると、性能がどう変わるかをQM/MM(反応など詳細を把握する箇所は量子化学、それ以外は古典力学に基づく分子力学で評価する手法)計算で評価します。これにより、反応のエネルギー障壁を下げうる、より高性能な改変酵素の設計に役立てることができます。
- 基質の前処理設計:
- 酵素が効率よく働くには、プラスチックにアクセスしやすくしてあげる必要があります。粉砕して表面積を増やす、非晶質領域を増やすといった前処理が、酵素の反応性にどう影響するかについてシミュレーションを用いて分子レベルの傾向を評価し、実験条件検討の指針とします。
![図3. イデオネラ・サカイエンシスの2つの酵素によるPETの分解 [6][7]。PETaseとMHETase、2つの酵素の連携によりPET→MHET→テレフタル酸+エチレングリコールの化学反応が生じる。](/tech-blog/aluome00000000ej-img/003.jpg)
PETaseとMHETase、2つの酵素の連携によりPET→MHET→テレフタル酸+エチレングリコールの化学反応が生じる。
(CC BY Attribution 4.0 International ライセンスの下で一部改変して使用)
7. ビトリマーなど新素材を活用したリサイクル
一度固めると二度と形を変えられない従来の熱硬化性樹脂には、「リサイクルできない」という弱点があります。この課題を克服する材料として、ビトリマーが注目されています。ビトリマーは、加熱されるとネットワーク内の結合が可逆的に組み替わる「動的共有結合」を持つため、架橋構造を保ちながらも流動し、再成形することが可能です。なお、このように共有結合でできたネットワークのつながり方がゆっくり変化する材料は、総称して Covalent Adaptable Networks(CANs)と呼ばれ、その代表例の1つがビトリマーです。
このユニークな挙動を整理するためによく用いられる指標が、トポロジー凍結転移温度(Tv)です。Tvは、結合交換反応が実質的に停止する(ネットワーク構造が凍結する)温度を指し、通常はレオロジー測定などの実験から評価されます。このTvを上回る温度に加熱すると、結合の組み替えが活発になり、材料の粘度が劇的に低下して加工しやすくなります[8]。
分子動力学シミュレーション(全原子モデル/粗視化モデル)を用いることで、温度領域ごとに、分子やネットワークがどの程度動くのか、結合のつなぎ変えがどのくらい進むのかといった「ふるまい」を調べます。シミュレーションを用いることで、実験前に温度と分子の運動性の関係などをおおよそ把握し、目的の加工条件に合ったビトリマーの材料選定や設計の指針を得ることができます[8]。
![図4.CANs(Covalent Adaptable Networks)の種類 [8]。 解離型(dissociative)と会合型(associative)に分類され、ビトリマーは後者の代表例。](/tech-blog/aluome00000000ej-img/004.jpg)
解離型(dissociative)と会合型(associative)に分類され、ビトリマーは後者の代表例。
(CC BY Attribution 4.0 International ライセンスの下で使用)
8. おわりに
本記事でご紹介した量子化学から連続体までのマルチスケールシミュレーションや、データサイエンスとの組み合わせによる条件絞り込みは、高度な連携が求められます。こうした統合的なアプローチは、たとえば「J-OCTA」のような統合支援システムを用いることで、よりスムーズなワークフローとして実行可能です。もしご関心をお持ちでしたら、お気軽にご連絡ください。
- van den Tempel, P., Picchioni, F., Recycling 2025, 10, 1.
(https://doi.org/10.3390/recycling10010001) - https://www.jsol-cae.com/product/material/jocta/feature/polymeric_material_simulation.html
- Deng,L., et al., Polymers, 2021,13, 3783.
(https://doi.org/10.3390/polym13213783) - Clark, R. A., Shaver, M. P., Chem. Rev. 2024, 124, 2617–2650.
(https://doi.org/10.1021/acs.chemrev.3c00739) - Mieda, S., ACS Omega 2025, 10, 5973–5980.
(https://doi.org/10.1021/acsomega.4c09953) - Knott, B. C., et al. PNAS 2020, 117, 25476–25485.
(https://doi.org/10.1073/pnas.2006753117) - dos Santos, A. M., et al., J. Phys. Chem. B 2024, 128, 7486–7499.
(https://doi.org/10.1021/acs.jpcb.4c02207) - Karatrantos, A. V., et al., Polymers 2024, 16, 1373.
(https://doi.org/10.3390/polym16101373) - Peti, D., et al., Polymers 2025, 17, 603.
(https://doi.org/10.3390/polym17050603)
技術ブログカテゴリ
新着記事
- IMPETUS/VALIMATユーザー会2026を開催しました
- 接着・粘着設計とマルチスケールシミュレーション
- 医療画像3Dモデリングとは?CT・MRI(DICOMデータ)から3Dモデルを作る方法
- 微細化時代における半導体設計1:微細化と設計指針の変化
- 塗膜乾燥のシミュレーション — 塗布・塗装・電極プロセスの1D/3D解析入門
- マテリアルズ・インフォマティクス(MI)とは?
AI×シミュレーションで加速する次世代材料開発 - リアルワールドの自動車衝突安全に向けて(2)~ISOレーティング~
- 分子シミュレーションで設計するプラスチックリサイクル
- 形状設計フェーズでの組み立て精度向上によるコスト削減
~ 組み立てCE検討ツールのご紹介 ~ - JSOLが考える「溶接シミュレーションと工場デジタルツインが実現する工程設計」について講演しました
