
マルチスケールシミュレーション・ソフトウェア
J-OCTA
マルチスケールシミュレーション・ソフトウェア
J-OCTA
金属表面と分子の相互作用
J-OCTAのSIESTA界面エネルギー計算ツールを用いて、銅(111)表面に吸着するアルカン分子との相互作用を解析しました。第一原理計算である密度汎関数理論(DFT)により得られた吸着エネルギーをGeneralized Lennard-Jones関数でフィッティングし、得られたパラメータを用いて全原子分子動力学(Full-Atomistic MD)計算を実施しました。炭素数の増加に伴う吸着エネルギーの変化も再現されています。
解析・利用例のポイント
- J-OCTA SIESTA 界面エネルギー計算ツールの活用事例
- MDシミュレーションのためのパラメータの決定
- 無機-有機界面の様々な現象に適用可能
J-OCTA SIESTA界面エネルギー計算ツールの活用事例
銅(111)面に吸着するアルカン分子の様子が示されています。J-OCTAのSIESTA界面エネルギーツール(Interfacial Energy Tool)を用いて、有機-無機界面の力場パラメータを決定し、全原子分子動力学シミュレーションを行っています。
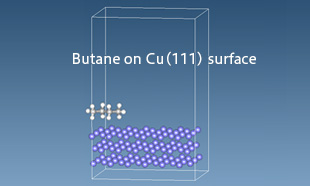
銅(111)面に吸着するアルカン分子
MDシミュレーションのためのパラメータの決定
アルカン分子(プロパン、ブタン、ペンタン、ヘキサン)と銅表面の相互作用エネルギーをSIESTAを用いたDFTで計算し、分子動力学(MD)で用いる力場パラメータをGeneralized Lennard-Jones関数でフィッティングした結果が示されています。
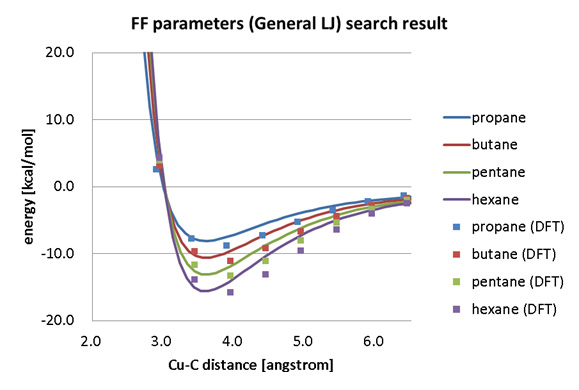
力場パラメータフィッティング結果
解析内容の詳細
関連情報
お役立ち資料

ソリューション

材料シミュレーションとマテリアルズ・インフォマティクス(自動車)
データとシミュレーションを連携した材料開発(マテリアルDX)
材料シミュレーションとマテリアルズ・インフォマティクス(輸送機器)
データとシミュレーションを連携した材料開発(マテリアルDX)

材料シミュレーションとマテリアルズ・インフォマティクス(化粧品/食品)
シミュレーションとデータサイエンスの連携による材料開発(マテリアルDX)

シミュレーションとデータサイエンスの連携による医薬品開発(医薬品)
創薬と製剤分野への先端シミュレーション技術とAIの適用

材料シミュレーションとマテリアルズ・インフォマティクス(化学/材料)
シミュレーションとデータサイエンスの連携による材料開発(マテリアルDX)

半導体デバイスの材料シミュレーションとマテリアルズ・インフォマティクス
シミュレーションとデータサイエンスの連携による半導体デバイス材料開発(マテリアルDX)
セミナー・イベント

受付中
体験セミナー
材料設計のためのマルチスケールシミュレーションセミナー
日程:定期開催
会場:開催形式:WEB

受付中
体験セミナー
第一原理計算を用いた材料設計セミナー(SIESTA/Quantum ESPRESSO)
日程:定期開催
会場:開催形式:WEB

受付中
CAEトレーニング
マテリアルズ・インフォマティクスの基礎と実践セミナー(Data-driven材料設計の基礎と実践)
日程:定期開催
会場:開催形式:WEB

受付中
CAEトレーニング
ライフ/バイオ分野のためのマルチスケールシミュレーションセミナー
日程:定期開催
会場:開催形式:WEB

受付中
CAEトレーニング
一歩進んだJ-OCTA利用のためのPythonスクリプトセミナー
日程:定期開催
会場:開催形式:WEB

受付中
CAEトレーニング
材料同士の親和性評価のためのソリューションセミナー
日程:定期開催
会場:開催形式:WEB
技術ブログ

高分子材料シミュレーションの概要と事例
高分子のシミュレーション技術について代表的ないくつかの手法の概要、それぞれの連携、ソフトウェアなどを紹介します。
2022.05.01
【中級編】シミュレーション活用ナレッジ
製造業におけるマテリアルズ・インフォマティクス
マテリアルズ・インフォマティクス(MI)が注目されてから10年以上経ちました。多くの企業でMIの導入と活用が進められていますが、課題も見えて...
2025.05.29
【中級編】シミュレーション活用ナレッジ
ライフサイエンス/バイオマテリアル分野のための分子シミュレーション
ライフサイエンス/バイオマテリアル分野の製品機能最適化に重要な分子シミュレーションの活用例をご紹介
2024.08.01
【中級編】シミュレーション活用ナレッジ
