
マルチスケールシミュレーション・ソフトウェア
J-OCTA
マルチスケールシミュレーション・ソフトウェア
J-OCTA
MDおよびMO/DFTを用いた比誘電率の評価
分子動力学(MD)と量子化学計算(MO/DFT)を用いて、ベンゼン・アセトン・PVCなどの比誘電率を評価しました。MDでは双極子モーメントの時間揺らぎから配向分極を、MO/DFTでは分極率から電子分極を算出しました。実験値との比較により、分子構造に応じた誘電率の寄与を明らかにします。
解析・利用例のポイント
- 様々な分子の比誘電率の評価
- 誘電率の起源について理解可能な解析
- ポリマーへの適用可能性
様々な分子の比誘電率の評価
ベンゼンとアセトンの解析モデルが示されています。MDおよびMO/DFTを用いて比誘電率の評価を行うための構造が視覚的に示されています。
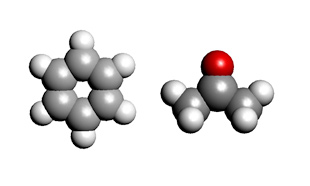
解析モデル(左:ベンゼン、右:アセトン)
誘電率の起源について理解可能な解析
ベンゼンとアセトンに対してMOとMDによって算出された比誘電率の結果が示されています。MDはOPLS力場を用い、MOはGaussianによる分子分極率を使用しています。
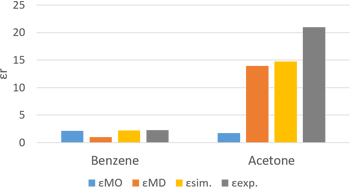
比誘電率計算結果
ポリマーへの適用可能性
PVCポリマーの比誘電率をMDおよびQSPRにより評価した解析モデルが示されています。MDでは100分子系を用い、QSPRでは密度から推算しています。
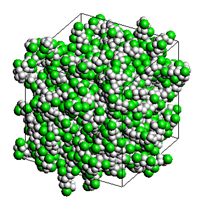
解析モデル(PVC)
参考文献
- J.Bicerano, Prediction of Polymer Properties, 3rd Ed. Marcel Dekker, 2002
- J. Chem. Eng. Data 2018, 63, 5, 1170
関連情報
お役立ち資料

ソリューション

材料シミュレーションとマテリアルズ・インフォマティクス(自動車)
データとシミュレーションを連携した材料開発(マテリアルDX)
材料シミュレーションとマテリアルズ・インフォマティクス(輸送機器)
データとシミュレーションを連携した材料開発(マテリアルDX)

材料シミュレーションとマテリアルズ・インフォマティクス(化粧品/食品)
シミュレーションとデータサイエンスの連携による材料開発(マテリアルDX)

シミュレーションとデータサイエンスの連携による医薬品開発(医薬品)
創薬と製剤分野への先端シミュレーション技術とAIの適用

材料シミュレーションとマテリアルズ・インフォマティクス(化学/材料)
シミュレーションとデータサイエンスの連携による材料開発(マテリアルDX)

半導体デバイスの材料シミュレーションとマテリアルズ・インフォマティクス
シミュレーションとデータサイエンスの連携による半導体デバイス材料開発(マテリアルDX)
セミナー・イベント

受付中
体験セミナー
材料設計のためのマルチスケールシミュレーションセミナー
日程:定期開催
会場:開催形式:WEB

受付中
体験セミナー
第一原理計算を用いた材料設計セミナー(SIESTA/Quantum ESPRESSO)
日程:定期開催
会場:開催形式:WEB

受付中
CAEトレーニング
マテリアルズ・インフォマティクスの基礎と実践セミナー(Data-driven材料設計の基礎と実践)
日程:定期開催
会場:開催形式:WEB

受付中
CAEトレーニング
ライフ/バイオ分野のためのマルチスケールシミュレーションセミナー
日程:定期開催
会場:開催形式:WEB

受付中
CAEトレーニング
一歩進んだJ-OCTA利用のためのPythonスクリプトセミナー
日程:定期開催
会場:開催形式:WEB

受付中
CAEトレーニング
材料同士の親和性評価のためのソリューションセミナー
日程:定期開催
会場:開催形式:WEB
技術ブログ

高分子材料シミュレーションの概要と事例
高分子のシミュレーション技術について代表的ないくつかの手法の概要、それぞれの連携、ソフトウェアなどを紹介します。
2022.05.01
【中級編】シミュレーション活用ナレッジ
製造業におけるマテリアルズ・インフォマティクス
マテリアルズ・インフォマティクス(MI)が注目されてから10年以上経ちました。多くの企業でMIの導入と活用が進められていますが、課題も見えて...
2025.05.29
【中級編】シミュレーション活用ナレッジ
ライフサイエンス/バイオマテリアル分野のための分子シミュレーション
ライフサイエンス/バイオマテリアル分野の製品機能最適化に重要な分子シミュレーションの活用例をご紹介
2024.08.01
【中級編】シミュレーション活用ナレッジ
