
リチウムイオン電池のシミュレーション
全固体電池の実用化時期が2020年代後半以降にずれ込むとの見通しから、EV向けバッテリの研究・開発は、候補となる材料の可能性を広く探る方向にシフトしています。このため、全固体電解質を含めた幅広い材料探索の観点から、材料系シミュレーションに対する期待とニーズが高まってきています。
本稿では、リチウムイオン電池の構成要素である負極/正極/電解質と製造工程を対象として、シミュレーションによる評価・検討事例を紹介します。
1.負極解析
リチウムイオン電池は、吉野博士が当時正極材料として知られていたLiCoO2に対する負極材料として、炭素材料を見出したことが実用化の原動力になったことはよく知られています。負極に使用される炭素材料は、黒鉛層間物質と呼ばれる層状炭素にリチウムをドープした化合物を利用します。 実用上は充電によりLiCoO2からLiイオンを脱離させ、層状炭素にLiイオンをドープすることで、黒鉛層間物質であるLiC6が生成されます(放電時はこの逆過程)。この過程は、理論的には充放電に伴うLiイオンの正負極間における可逆過程ですが、Liイオンの吸蔵と脱離により、電極では体積の膨張と収縮、弾性率の変化を生じることが予想されます。
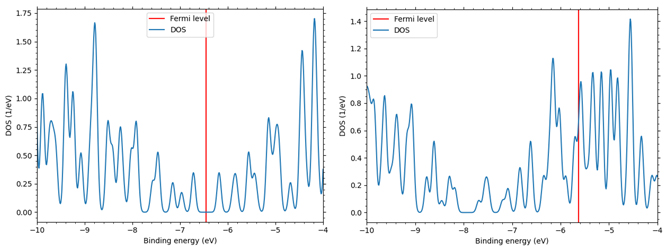
この様子を第一原理計算エンジンであるSIESTAにより解析を行い、確認しました。図1に緩和解析前の充電後のLiC6としてLiイオンがグラファイトにドープされた状態のモデル図(左)と放電後のLiイオンが完全に脱離したと仮定したときのグラファイトのみのモデル図(右)を示しました。表1は、図1のモデルに対して緩和解析を行い、層間長、体積変化、層間結合の強さを表すc33を示しています。この結果から、Liイオン脱離後の層間長と体積を1とすると、Liイオン吸蔵時には、層間長で約8%、体積で約10%の膨化が見られることが分かりました。また相関結合の強さを示すc33は、LiC6にくらべてグラファイトでは40%ほど低減していることが分かります。さらに両者の電子状態の違いをそれぞれの状態密度としてみた結果を図2に示します(赤線はフェルミ準位)。図2において、左はLiC6の状態密度で右が黒鉛層の状態密度です。両者のフェルミ準位の違い(ギャップ状態からギャップレス状態へ変化)からグラファイトに比べてLiC6は金属的な傾向が見られることが分かりました。

表1 グラファイトとLiC6の格子定数とC33の比較
| 層間距離(Å) |
層間距離比 グラファイト基準 |
体積比 グラファイト基 |
C33(GPa) | |
| グラファイト | 3.48 | - | - | 38.7 |
| LiC6 | 3.74 | 1.08 | 1.10 | 60.5 |
リチウムイオン電池の特性向上を目指した新しい負極材料の探索と研究も盛んに行われています。リチウムイオン電池の容量は負極容量で決まることから、Liを多く吸蔵できる炭素系以外の材料も検討されてきました。 Siから構成される負極はこれまでの炭素材料による負極に比べて、理論的には10倍以上の容量を持つことが知られています。しかしSi負極はLiイオン吸蔵時の体積膨張率が400%と、極めて大きな膨化現象を示すため、体積変化による負極の崩壊が開発上の大きな課題となっており、シミュレーションを利用した負極崩壊のミクロなメカニズムの解明が期待されます。
文献[1]では、SIESTAによる第一原理計算により、アモルファスSiから構成された負極上のLiの吸蔵と脱離の過程において生じる応力と歪みの関係や、Si負極原子のリチウム化(金属化)が報告されています。この解析では、Liの濃度を外部の化学ポテンシャルとして制御することで、アモルファスSiへの吸蔵過程と脱離過程をシミュレーションしています。解析の結果、横軸をLi濃度としたときの応力履歴は、ヒステリシスを描いており、Liの吸蔵と離脱の過程において不可逆変化を生じていることが分かりました。力学特性の観点からは、Si負極にLiが入り込むことによる負極原子のリチウム化のため、ヤング率などの低下が確認されています[2]。Liの吸蔵は、膨化に見られる形状変化だけでなく、Si-Si結合の切断とSi-Li生成をもたらします。電子論的には、Si負極の半導体状態から金属状態への遷移であることがフェルミ準位における状態密度の変化を通して確認されています[1]。
2.正極解析
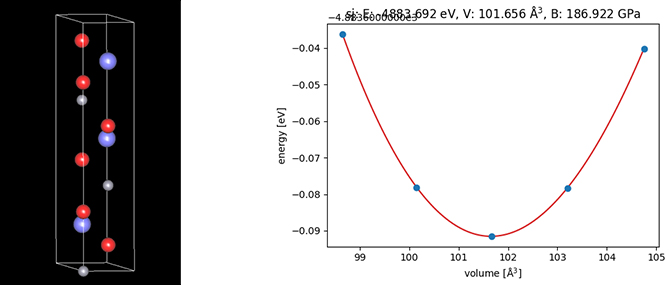
正極内の活物質においても、Liイオンの吸蔵と脱離により弾性率の変化が生じることが予想されます。シミュレーションにおいては、たとえばLi1-xCoO2のヤング率、体積弾性率などの力学特性が第一原理計算により求められており[3][4]、これらの結果によれば、Liイオンが完全に脱離したCoO2(x=1)における体積弾性率がLiCoO2(x=0)のそれにくらべて、半分程度になることが報告されています。シミュレーションでは、Liの割合を示す1-xの値がもっとも簡単な整数比になるように、Li1-xCoO2の単位セルを複数あわせたスーパーセルを定義して解析します。
第一原理計算エンジンSIESTAのモデラに含まれるEOS(状態方程式)機能を利用して、体積弾性率を算出しました(図3)。物理的には結晶のスティフネステンソルを求めることで、ヤング率とポアソン比、あるいは体積弾性率と剪断弾性率を求めることができます。シミュレーションにおいては、歪みを加えた後の形状最適化計算(緩和計算)を行って得られる応力や弾性エネルギーの解析を通して、これらの値を求めることができます。
3.電解質解析
全固体電池として現在研究されている電解質には、大きく硫化物系、酸化物系、ポリマー系の3種類に分類されます。現状では硫化物系電解質のイオン伝導度が最も高く、実用化に近いと考えられてきました。しかしEV向けバッテリにおいて、Liイオンの吸蔵/脱離に伴う体積変化による電極界面での劣化が問題となり、実用化は2020年代後半からとみられています。このため、イオン伝導度では硫化物系に及びませんが、酸化物系の検討も進められています。
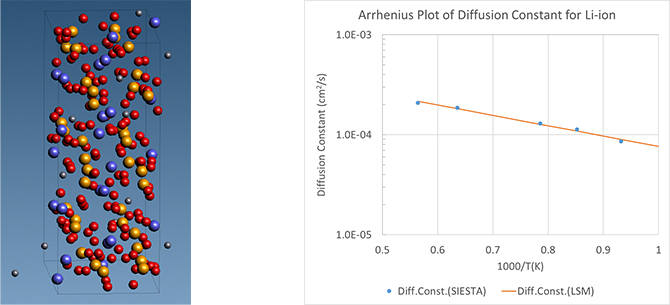
検討されている候補のひとつに、NASICON型に属するLiZr2(PO4)3(LZP)があります。イオンの移動度は常温域におけるネルンスト-アインシュタインの関係式を通して、拡散定数から求めることができます。J-OCTAのSIESTAモデラを用いて、ユニットセルが菱面体晶系であるα型結晶6セル分から構成されるスーパーセルを構築しました。SIESTAを用いた第一原理MD計算を実施し、LZPの拡散定数を求めた結果をアレニウスプロットとして、図4に示しました。文献[5]との比較のため、温度範囲を673~1773Kとしています。計算の結果、1000K以上の領域では、定量的にも文献[4]と一致する傾向が見られました。他方で1000K未満の領域では、解析の温度範囲は1073K~1773Kとしています。低温域での拡散定数の見積もりを行うには1000K以下の温度範囲も加えるべきですが、今回の解析時間内では、1000K以下の温度範囲において、異常拡散効果が観測され、十分な線形域が採れなかったため、結果から外しています。解析時間の違い(文献[5]のおよそ1/10の解析時間)と1000K以上の高温域のため、文献[5]との直接の比較はできませんが、類似した傾向が見られています。
なお、拡散定数のシミュレーションによる見積もりについては、短時間のシミュレーション結果から,機械学習によって長時間シミュレーションの結果を予測する「MD-GAN」[6]が有効であることが示されています。特に第一原理MDでは長時間のダイナミクスの予測は困難であることから、今後の主要な解析手法の一つになると考えられます。
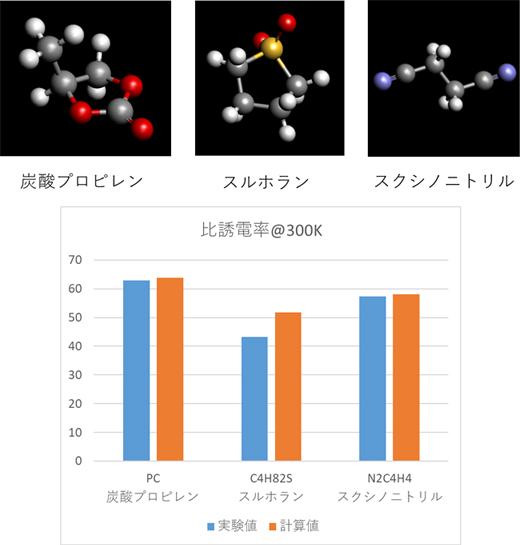
またLi塩濃度がこれまでの液系リチウムイオン電池に比べて数倍におよぶ高濃度のLi塩を溶解させるための液系溶媒の研究も進められています。高濃度のLi塩を溶解するには、Liイオンの電離を促進する比誘電率の高い溶媒が求められます。このような溶媒として、炭酸プロピレン、スルホラン、スクシノニトリルなどが挙げられます。これらの比誘電率は、分子内の電気双極子モーメントと原子内の電子分極により特徴づけられます。
これらの寄与をJ-OCTAの分子動力学シミュレーションとGaussianなどの分子軌道計算により算出し、Onsager/Kirkwoodの式により比誘電率を評価した結果を図5に示します。
4.生産工程解析
リチウムイオン電池の製造工程において、核となる技術のひとつにスラリー塗工のプロセスがあります。塗工時の正負極の膜厚やその膜厚比によってバッテリの容量/レート特性などに影響を及ぼすため、非常に重要なプロセスとされています。塗工プロセスでは、集電体となる金属箔に活物質、バインダー、有機溶媒なとからスラリー(固体粒子が懸濁した流動体)を塗工し、その後乾燥処理により溶媒分子を蒸発させます。
文献[7]では、J-OCTAに含まれる粗視化分子動力学法を利用して、活物質、バインダー、有機溶媒を粗視化したモデルを作成し、有機溶媒の蒸発過程をシミュレーションしました。
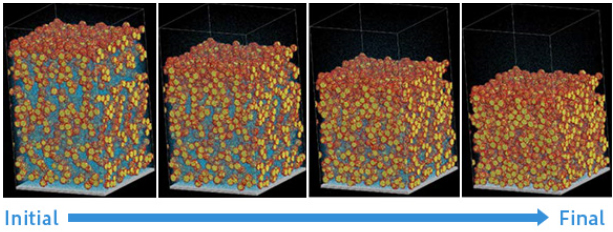
図6に溶媒分子の蒸発による塗工膜の時間変化の様子を示します。 時間の経過と共に、表面に近い領域から溶媒分子の蒸発が進み、多孔質構造が形成されていることが分かります。
5.おわりに
バッテリ内で生じる現象には、たとえばSi負極における膨化現象のように目で見てはっきりと分かるものもあります。しかしその起源を辿ってゆくとSi負極のリチウム化のように電子スケールでの物理的な変化が関わっており、マルチスケールな物理現象と言えます。J-OCTAはバッテリ内に生じるマルチスケールな現象を様々な視点からマルチスコープ的に捉えたシミュレーション技術を提供しています。
- Kejie Zhao, Georgios A. Tritsaris, Matt Pharr, Wei L. Wang, Onyekwelu Okeke, Zhigang Suo, Joost J. Vlassak, and Efthimios Kaxiras, Nano Lett. 2012, 12, 4397-4403
- Kejie Zhao, Wei L. Wang, John Gregoire, Matt Pharr, Zhigang Suo, Joost J. Vlassak, and Efthimios Kaxiras, Nano Lett. 2011, 11, 7, 2962-2967
- Yue Qi, Louis G. Hector, Jr., Christine James, and Kwang Jin Kim, Journal of The Electrochemical Society, 161 (11) F3010-F3018 (2014)
- Linmin Wu and Jing Zhang, JOURNAL OF APPLIED PHYSICS 118, 225101 (2015)
- Yusuke Noda, Koki Nakano, Hayami Takeda, Masashi Kotobuki, Li Lu, and Masanobu Nakayama, Chem. Mater. 2017, 29, 8983-8991
- K. Endo, K. Tomobe and K. Yasuoka. Proc. Conf. AAAI Artif. Intell., 2018.
- 諸星圭、土橋利幸、河村芳海、茶木健太、大畠広介、小沢拓、HPCI Research Report Vol.3 (2018) 89-94
- ※ 記載されている製品およびサービスの名称は、それぞれの所有者の商標または登録商標です。
この記事の関連情報
技術ブログカテゴリ
新着記事
- 塗膜乾燥のシミュレーション — 塗布・塗装・電極プロセスの1D/3D解析入門
- マテリアルズ・インフォマティクス(MI)とは?
AI×シミュレーションで加速する次世代材料開発 - リアルワールドの自動車衝突安全に向けて(2)~ISOレーティング~
- 分子シミュレーションで設計するプラスチックリサイクル
- 形状設計フェーズでの組み立て精度向上によるコスト削減
~ 組み立てCE検討ツールのご紹介 ~ - JSOLが考える「溶接シミュレーションと工場デジタルツインが実現する工程設計」について講演しました
- リアルワールドの自動車衝突安全に向けて
- 機械学習で加速する材料シミュレーション技術
- ノウハウ不要!樹脂の複雑な材料特性を簡単にフィッティング 〜 材料同定ツールと高精度ユーザーサブルーチンの活用事例 〜
- 樹脂材やゴム材の高精度予測に向けたパラメータ同定
