


[解析事例]表面の再構成
- 量子化学・DFT
- 界面・相分離・粒子分散性
- マテリアルサイエンス
Siの表面構造の計算
固体表面は、表面を構成する原子が周囲原子との結合を失うことでエネルギー的にバルク状態よりも不利な状況になります。エネルギーの損失の大きさは表面エネルギーとして知られており、これは面方位によって異なる値を持ちます。(参照:[解析事例] 顔料の表面エネルギー)
また、表面付近では電子状態がバルクとは異なるため、金属などの表面付近の面間隔は結晶内部とはずれることが知られています。
共有結合性の固体では、1次元的な構造緩和だけでなく面内での2次元的な緩和が起こることが知られています。Si(001)面は表面の原子がダイマー化することが知られています。
ここではSi(001)面の表面構造の緩和について、第一原理MD計算で検討しました。
SIESTAモデラのMD計算機能を使ってSi(001)面のMD計算を行いました。汎関数はPBE、基底関数はSZPを用いました。時間ステップは2fsで計算しトータル1psの計算を行いました。この計算では表面付近3層の原子を除き、拘束しています。図1にはMD計算中のポテンシャルエネルギーをプロットしています。0.8psのスナップショットを見ると、ダイマー構造が形成されているのが確認できます。ポテンシャルエネルギーの変化を見ると、いくつかのエネルギーの谷構造を超えており、単なる最適化計算ではダイマー構造に到達しない可能性があります。
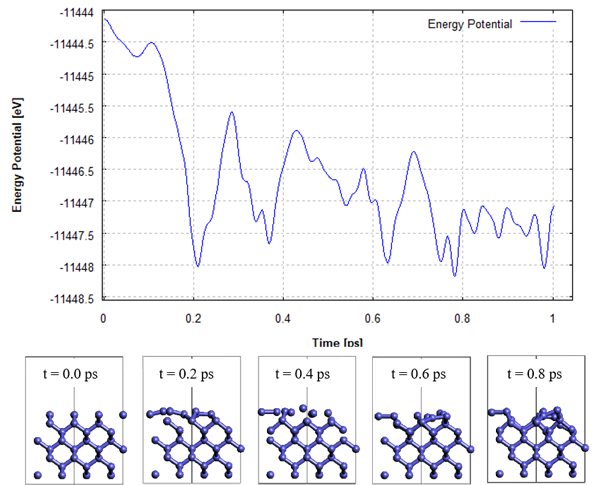 図1 Si(001)面の表面再構成の第一原理MD計算
図1 Si(001)面の表面再構成の第一原理MD計算
SiO2の表面構造の計算
同様の計算例として、シリカのQ4表面の作成を第一原理MDで行った例を紹介します。
Q4表面は表面酸素がすべてSi原子と結合している表面であり、疎水的な表面として知られています。この表面は特徴的な環構造を有しています。
MD計算はクオーツの構造を初期構造として用いています。時間ステップ2fsで200ステップ計算を行ったところ、目的の構造が得られました。
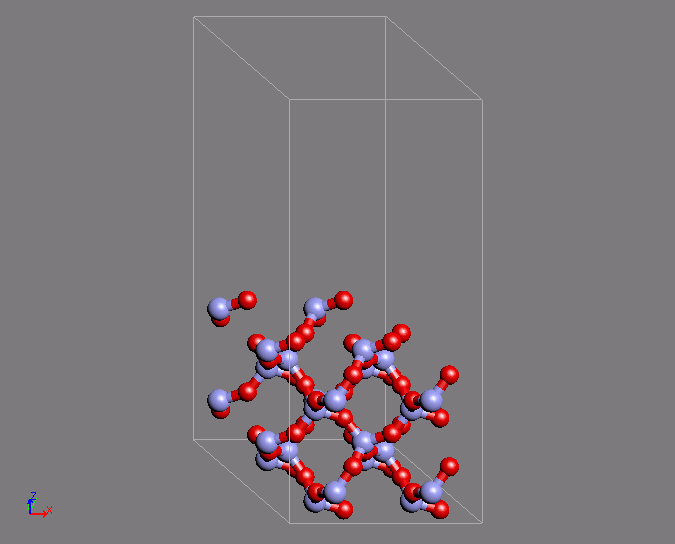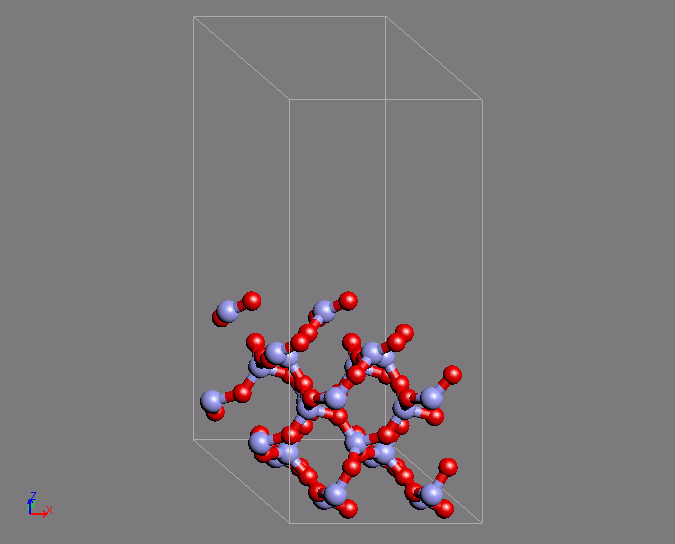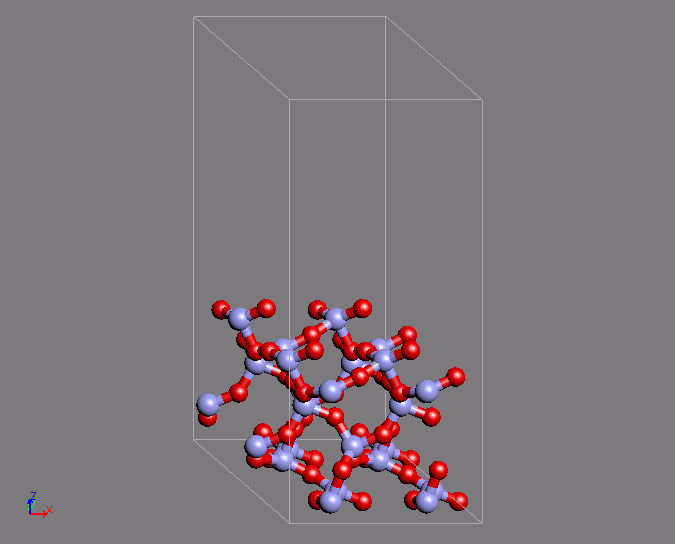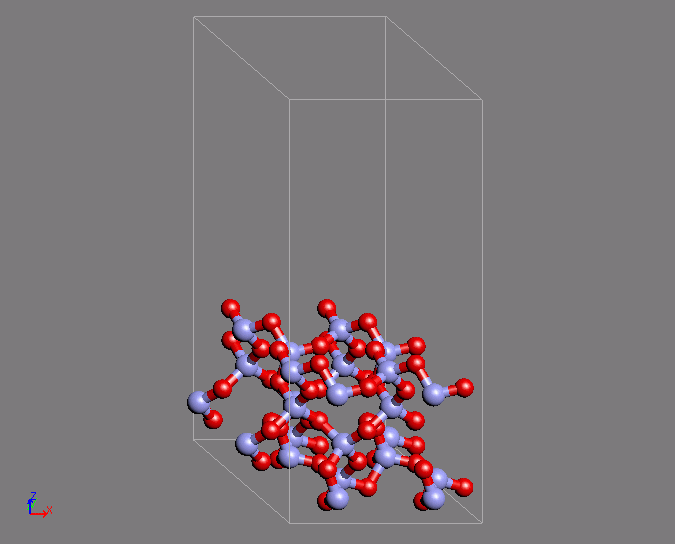
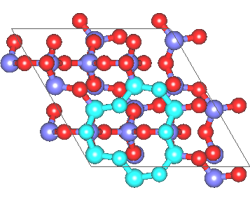
図2 第一原理MD計算によるシリカQ4表面構造の形成(左)初期構造(右)表面の特徴的な環構造
事例一覧
- ※記載されている製品およびサービスの名称は、それぞれの所有者の商標または登録商標です。
