


[解析事例] 金属表面と分子の相互作用
- 量子化学・DFT
- 全原子MD
- マルチスケール解析
- 低分子の浸透・拡散・吸着
- 界面・相分離・粒子分散性
- マテリアルサイエンス
目的と手法
J-OCTAのSIESTA界面計算ツールを用いることで、有機-無機界面で用いる力場パラメータを構築し、界面の分子動力学シミュレーションを行うことが可能です。
図1には銅(111)面に吸着するアルカン分子の様子が示されています。J-OCTAではこのような表面のモデリングと、表面と分子の相互作用計算を簡便に実施することが可能です。
 図1. 銅(111)面に吸着するアルカン分子
図1. 銅(111)面に吸着するアルカン分子
解析結果
図2はアルカン分子(プロパン、ブタン、ペンタン、ヘキサン)と同表面の相互作用エネルギーのDFTによる計算結果と、Generalized Lennard Jones関数を用いてパラメータフィッティングを行った結果です。
フィッティングでは以下の式で定義する差δEiの平均二乗誤差を最小化することを行いました。
![]()
フィッティングしたパラメータはすべての分子で共通のものを用いているものの、DFTの結果をおおむね再現しており、炭素数の増加に対する吸着エネルギーの変化もよく再現しています。
得られたパラメータを用いて、デカン分子と銅表面のMD計算に適用した結果が動画です。
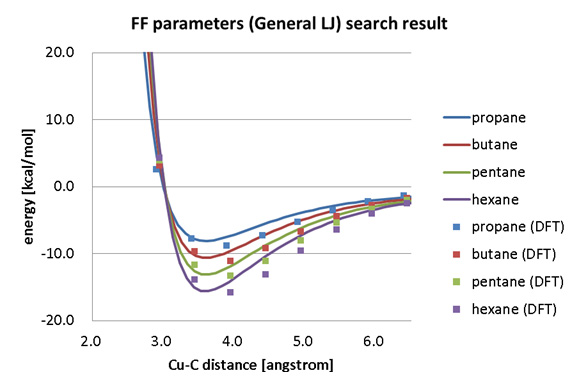 図2. FFパラメータフィッティング結果
図2. FFパラメータフィッティング結果
 動画:デカン分子と銅表面のMD計算 パラメータ適用結果
動画:デカン分子と銅表面のMD計算 パラメータ適用結果

このような十分な大きさの表面積を有するスラブモデルを計算する場合、100以上の原子数の設定が必要であり、膨大な計算時間を要することが多いですが、SIESTAを用いることで一般的なワークステーションでも高速に計算を実施できます。
事例一覧
-
- 機械学習ポテンシャルを用いた格子熱伝導率計算
- Martini3モデルによるナフィオン膜の計算
- 機械学習によるσプロファイルを記述子とした物性推算
- 液膜蒸発乾燥シミュレーション
- タイヤの耐摩耗性の向上
- COSMO法による物性推算
- VSOP-PSによる繊維配向材のシミュレーション
- 粗視化モデルによる脂質膜の解析
- 機械学習による沸点、屈折率、比誘電率の推算
- ナノトライボロジー(アブレシブ摩耗、ナノ加工)
- コバルト酸リチウムの基底状態と弾性率の解析(SIESTA事例ページへ)
- MD-GANによる固体電池内のLiイオン拡散解析
- リバースマッピングによるアモルファス構造の作成
- 電池電極の成形プロセス(カレンダリング)における圧力と空隙率の計算
- 粗視化分子動力学を用いた複屈折の解析
- 高分子膜の相分離プロセスシミュレーション
- 機械学習によるχパラメータの推定
- 水への溶解性評価
- フィラー樹脂複合材料の熱伝導率計算
- VSOP-PSによる繊維構造への樹脂含浸プロセス計算
- mol-inferを用いたQSPRの逆解析
- FMO-DPDを用いた高分子電解質の相分離構造
- FMO-DPDを用いた脂質膜とベシクルの形成
- カルサイト(方解石)の複屈折と光吸収(SIESTA事例ページへ)
- リチウムイオン電池のシミュレーション(SIESTA事例ページへ)
- 懸濁液の粘度の評価
- 量子補正を適用した固体の定積比熱の評価
- MD-GANを用いた長時間の分子運動の予測
- ポリマーの誘電緩和
- 金属錯体の吸収スペクトル(SIESTA事例ページへ)
- ゼオライトとガス分子の相互作用(SIESTA事例ページへ)
- 蒸着膜のシミュレーション
- 機械学習QSPRとマテリアルズ・インフォマティクス
- シミュレーション結果を用いた粘弾性マスターカーブの作成
- DPDを用いた粘弾性のシミュレーション
- 表面の再構成(SIESTA事例ページへ)
- フォノン分散を利用した剛性マトリクスの算出(SIESTA事例ページへ)
- Steered MDによるポリペプチドの自由エネルギー変化の解析
- 金属の電子比熱解析(SIESTA事例ページへ)
- 格子比熱の解析(SIESTA事例ページへ)
- J-OCTAによる蓄熱材の評価
- MDおよびMO/DFTを用いた比誘電率の評価
- 溶解度係数の算出
- 機械特性評価(SIESTA事例ページへ)
- 結晶の熱膨張(SIESTA事例ページへ)
- ゼオライトへのガス吸着
- GHz周波数領域における水の誘電分散(2)
- 反応のエネルギー変化(SIESTA事例ページへ)
- GHz周波数領域における水の誘電分散(1)
- 固体表面への分子の吸着エネルギー(SIESTA事例ページへ)
- 活性化エネルギーを用いたモンテカルロ判定によるエポキシ樹脂の架橋反応
- 樹脂 複屈折のためのマルチスケールシミュレーション
- 顔料の表面エネルギー(SIESTA事例ページへ)
- スラリー塗工プロセス
- 金属表面と分子の相互作用
- フィラー充填ゴムの繰り返し伸長
- 界面特性を考慮したCFRTPの破壊挙動の解析(Digimat事例ページへ)
- グラフェンシート添加によるCFRTPのマトリクス熱伝導特性改質(Digimat事例ページへ)
- 非平衡MDによる熱伝導率の計算
- ゴム材料のレオロジーシミュレーション
- 架橋フェノール樹脂の物性評価
- エポキシ樹脂架橋反応のシミュレーションからのガラス転移温度評価
- 電池用電解液の計算
- カーボンナノチューブの分散構造と非線形構造解析
- 粗視化ポテンシャルの評価
- DPDによる溶媒蒸発シミュレーション
- ガラス状態ポリマーの一軸伸長とクレーズ形成
- 高分子-固体界面の摩擦
- MDによる粘度の評価
- 相図からのχパラメータ推算
- 高分子-固体界面の剥離
- 気体の溶解係数と自由体積
- QSPRによる物性値の推算
- レオロジー特性
- 熱硬化性樹脂の架橋構造
- MDによる比誘電率の評価
- 複合材料の非線形力学特性(LS-DYNAとの連携)
- DPDによる界面張力の評価
- MDによる溶解度パラメータの評価
- DPDによる液滴のずり変形
- MDによる界面張力の評価
- 体積弾性率評価
- 界面活性剤の影響評価
- 燃料電池の高分子電解質膜解析
- 一軸伸張解析
- 光学特性評価
- ガラス転移温度評価
- ガス拡散解析
- ガス透過解析
- 架橋構造材料の特性評価
- コンポジット材料の特性解析
- ※記載されている製品およびサービスの名称は、それぞれの所有者の商標または登録商標です。
